
Quantification of subclonal selection in human cancers
Published:
This post will detail software used in our recent publication Quantification of subclonal selection in cancer from bulk sequencing data. For this publication we used a combination of mathematical and computational modelling, Bayesian inference and bioinformatics. We have tried to make the software/scripts for each of these components freely available (via github) and (hopefully) easy to use so that the results are as reproducible as possible and so that similar analyses can be performed with different datasets.
Frequentist based rejection of neutral evolution
As part of our study we first wanted to see what parameters of the evolutionary process in cancer we may feasibly be able to see in typical cancer sequencing datasets. Rather than go for the fully Bayesian approach we adopt later (which is very computationally expensive) we developed some new test statistics based on a model of neutral evolution we published previously (Williams et al. Nat Gen. 2016). These test statistics as well as some plotting functionality have been made available in an R package called neutralitytestr. A vignette describing how to use the package is available here.
As an example the package will produce figures like the following (which is a simulated dataset with a single subclone) and will also output values of the 4 test statistics we discuss in the paper with associated p-values generated empirically from a simulated cohort of tumours.
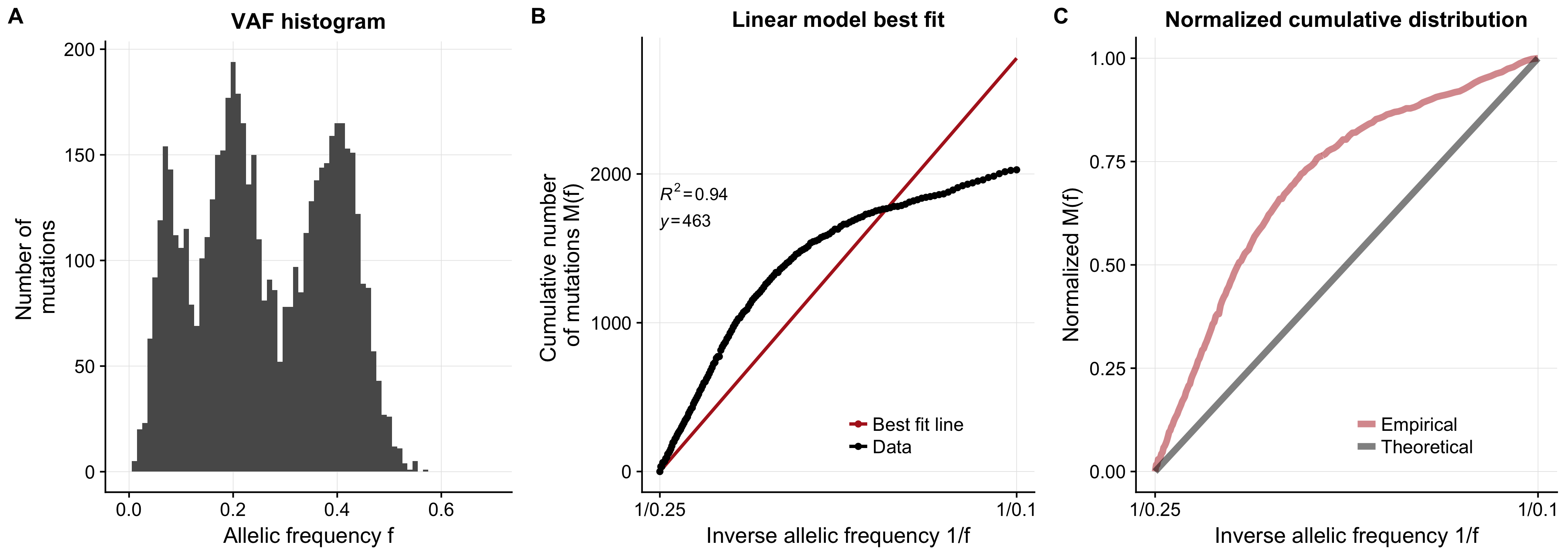
Using these statistics allowed us to asses the range of parameters we would expect to cause deviations from a neutral model, and the results are shown in Figure 1e of the paper.
Quantifying evolution in human cancers
The main results of the paper concentrate on attempting to measure evolution directly in human cancers from single samples at single time points. We developed a mathematical model of subclonal growth during tumour evolution and showed that using the variant allele frequency (VAF) distribution it is possible to measure several quantities that (using the mathematical model) enable us to infer the time subclones appear and their fitness. To measure these quantities we used a simulation of cancer evolution which incorporates sequencing noise together with approximate Bayesian computation, a technique which allows Bayesian inference to be performed using simulation based models.
We used the Julia programming language to implement this approach and the approach and idea is encapsulated in a package available here - SubClonalSelection.jl.
The package can output the model fits like the following plot which we used throughout the paper and posterior distributions for the parameters of interest.
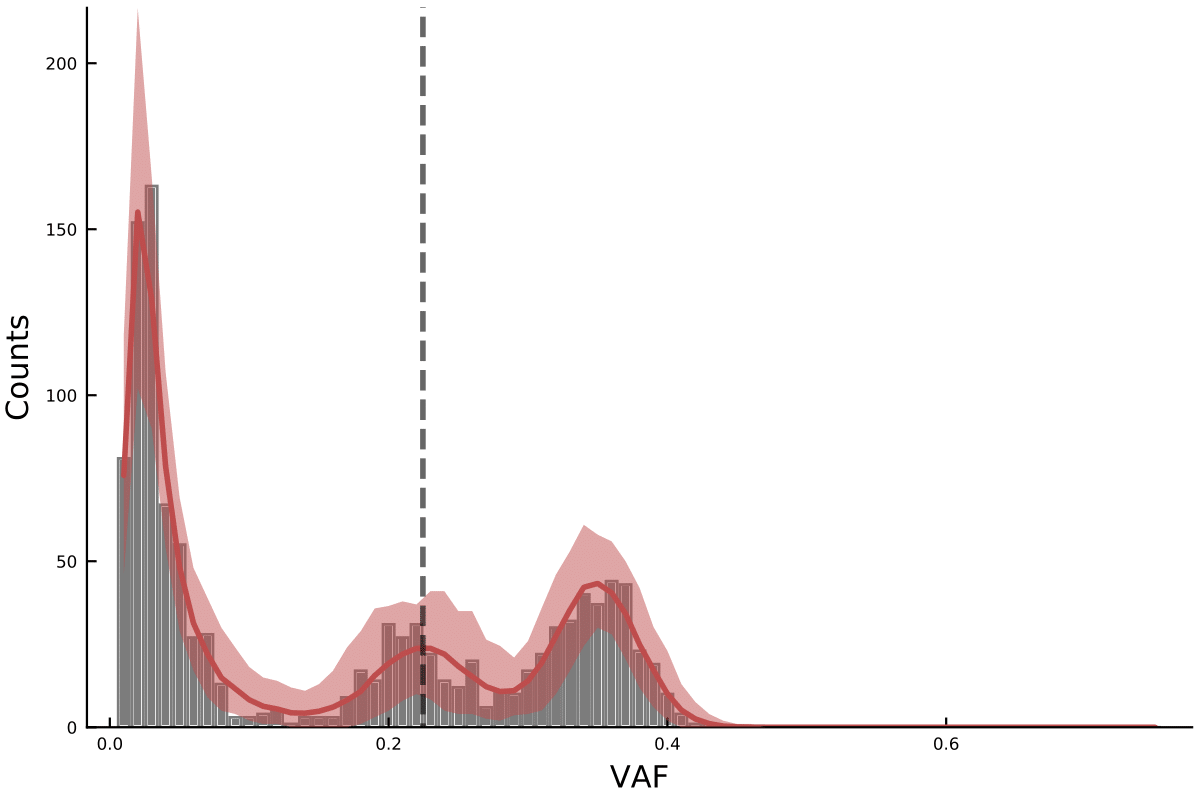
Detailed examples of how an analysis can be performed can be found here.
The above package uses ABC inference and a cancer evolution simulation which exist as standalone packages.
Bayesian inference
ApproxBayes.jl implements a simple rejection ABC approach and an ABC sequential monte carlo (ABC SMC) algorithm which is more efficient. Also included are model selection versions of both. In theory if you have your own simulation algorithm you could use it together with this package to do Bayesian inference with your model. It is fairly straightforward to use Julia with other languages (particularly C, C++ and python) so if you wanted to try out your own simulation it shouldn’t be too difficult.
Cancer modelling
CancerSeqSim.jl implements the computational model of cancer evolution. It uses a branching process model of cell divisions with mutations accruing at each division and then at the end implements an empirically motivated sampling procedure to produce synthetic “sequencing” data.
Bioinformatics
Where we were required to call mutations ourselves we used the Mutect2 algorithm for point mutations and sequenza for copy number calls. Scripts for this analysis are given here.
References
Williams, M. J., Werner, B., Barnes, C. P., Graham, T. A., & Sottoriva, A. (2016). Identification of neutral tumor evolution across cancer types. Nature Genetics, 48(3), 238–244. http://doi.org/10.1038/ng.3489
Williams, M. J., Werner, B., Heide, T. Curtis, C., Barnes, C. P., Graham, T. A., & Sottoriva, A. (2018). Quantification of subclonal selection in cancer from bulk sequencing data Nature Genetics http://dx.doi.org/10.1038/s41588-018-0128-6